昆华内分泌代谢专栏
本文来自云南省第一人民医院经典病例
作者:虞艳芳 审核:苏恒 编辑整理:周怡昆

云南省第一人民医院,又名“昆华医院”,其内分泌代谢科是云南省临床重点专科、糖尿病研究中心、继续医学教育基地,国家代谢性疾病临床医学研究中心核心成员单位,中国I型糖尿病联盟成员单位,中华医学会内分泌学分会全国委员、云南省医学会内分泌学分会的主任委员单位。
病史:患者 16 岁,女性。因原发性闭经入院。患者足月顺产,出生时身高体重不详,无产伤;自出生以来无头颅外伤史及高热感染史,身高及智力发育与同龄儿童无明差别,至今无乳房发育、无月经来潮。母亲诉,一直是女性外阴,入院后体检却发现短小阴茎。否认家族中有遗传病史。父母非近亲婚育,患者母亲(43 岁)及姐姐(23 岁)月经均正常,姐姐已婚生育一女,9 月大。
查体:身高:165 cm ,体重:47 kg,上部量:73 cm ,下部量:92 cm, BMI:17.3 kg/㎡, 体型稍偏瘦,面部皮肤多发痤疮,嘴唇毳毛多,喉结轻度显现。甲状腺未触及肿大。乳房 Tanner 分期Ⅰ期。阴毛浓密呈不规则形状,腋毛稀疏。双侧腹股沟区可扪及睾丸。外阴发育异常, 阴蒂肥大可见一大小约 2.0 cm×2.0 cm 突出物,呈小阴茎样, 质软,无压痛,尿道口与阴道口分开,妇科指检可探及阴道, 可容一指。
辅助检查:
染色体检查:46XY,患者 ZFX 基因和 ZFY 基因有扩增,SRY 基因有扩增。
甲状腺功能、肝肾功能及血电解质、血脂、血糖正常,立卧位血管紧张素Ⅰ(AⅠ)、血管紧张素Ⅱ(AⅡ)、肾素活性(PRA)、醛固酮(AID),17 α 羟孕酮(17α-OHP),生长激素(GH),24 h 尿 17-羟皮质类固醇(17-OH)、17-酮皮质类固醇(17-KS)、香草基杏仁酸(VMA)、动态皮质醇节律、硫酸脱氢表雄酮(DHEAS)、双氢睾酮(DHT)均正常。
雄烯二酮(AD)7.00 μg/L(男性参考值 0.3~3.5 μg/L)
血清睾酮(T)5.16(男性参考值 6.26~30.8 μg/L);
卵泡刺激素(FSH)20.26 IU/l(男性参考值 1.5~12.4 IU/l);
黄体生成素(LH)48.34IU/l(男性参考值 1.7~8.6IU/l);
雌二醇(E2)128.9pmol/l(男性参考值 94.8~223pmol/l)。
患者人绒毛膜促性腺激素(HCG)兴奋试验测睾酮和雄烯二酮无反应(雄烯二酮不反应,可能与患者隐睾,睾丸功能低下有关)
B 超显示:(1)先天性无子宫。(2)外阴实性低回声区(阴茎可能)。CT 检查结果:考虑为双侧隐睾;会阴部见刻意阴茎显示,形态短小;盆腔内未见确切子宫及双侧附件;双侧肾上腺未见明显异常。手腕骨龄片:正常。骨密度检查:正常。垂体磁共振成像(MRI)扫描:垂体增生。
讨论
性别可以分为染色体性别、性腺性别及表型性别,出现不一致时即为性分化异常。男性假两性畸形为性分化异常的常见疾患,其特点为内生殖器为男性 (睾丸、附睾),而外生殖器有不同程度女性化 (尿道下裂、阴唇、盲端阴道),原因有雄激素作用障碍以及雄激素合成障碍。
雄激素作用障碍包括雄激素受体基因 (AR) 突变导致的雄激素不敏感综合征及 5α-还原酶基因 (SRD5A2) 突变导致的 5α-还原酶缺陷症。雄激素合成缺陷常是由于肾上腺皮质类固醇激素合成过程中的酶缺陷导致胆固醇向雄激素转化障碍, 这些酶包括胆固醇侧链裂解酶、3β-羟类固醇脱氢酶 II 型、17α-羟化酶/17,20-裂解酶及 17β-HSD3, 其中 17β-HSD3 缺陷症最为常见。
本例患者虽社会性别是女性,但染色体为 46,XY;性腺为睾丸;有外生殖器男性化不完全表现。初步诊断考虑 46,XY 性分化异常(DSD)。患者无皮质醇合成障碍和肾上腺增生证据可排外先天性肾上腺增生症,双氢睾酮(DHT)正常可排外 5α还原酶缺陷症。该患者激素水平特点为促性腺激素水平显著高于正常而睾酮却低于正常,睾酮合成前体物质雄烯二酮明显升高。提示存在外周睾酮合成障碍。
进一步,hCG 兴奋试验显示睾酮/雄烯二酮比值未见升高,提示雄烯二酮转化成为睾酮过程受阻,因此考虑 17β-HSD3 缺陷症可能。为了进一步证实诊断,我们对本例患者及其家系进行了 HSD17B3 基因测序,患者(先证者)检测到两个基因变异: c.277+5 G>A; p.? 杂合,遗传自父亲;c.277 G>A; p.(Glu93Lys) 杂合,遗传自母亲。对其姐姐进行检测发现与患者相同,携带上述两个杂合基因突变,对患者外甥女进行上述两个位点基因检测,发现携带其中一个杂合突变:c.277 G>A; p.(Glu93Lys)
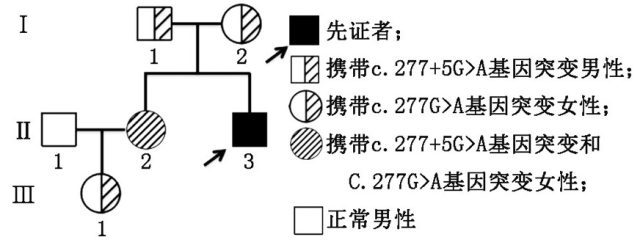
图 1 患者家系图
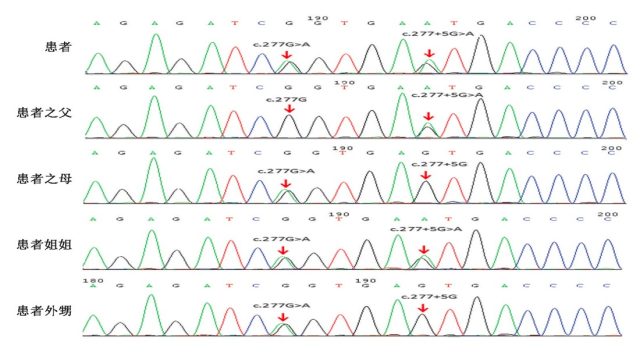
图 2 患者及其亲属 HSD17B3 基因测序结果
HSD17B3 基因位于染色体 9q22 区, 包含 11 个外显子。17β-HSD3 型缺陷症作为一种常染色体隐性遗传病,HSD17B3 基因的纯合突变或复合杂合突变均可致病 [1]。17β-HSD3 型主要在睾丸中表达,利用还原型辅酶Ⅱ(NADPH)作为辅因子催化雄烯二酮 (AD) 转化为睾酮(T), 对于胚胎期男性外生殖器的形成至关重要,所以该病主要影响男性性发育 [2]。
17β-HSD3 缺陷症的特征性临床表现是 46,XY 个体出生时内生殖器为睾丸, 外生殖器呈女性表型, 故常常被误按女性抚养, 青春期时却出现进行性男性化,表现为假两性畸形。17β-HSD3 缺陷症的致病基因至今已有 47 个突变被报道, 包括错义/无义突变、剪接突变、复制突变、插入、删失突变等 [3]。
在本例家系中, 患者有两个杂合突变点 c.277+5 G> A; p.?和 c.277 G>A; p.(Glu93Lys) ,分别各来自于其父母亲,患者的父母均为致病突变的携带者,但通常并不会发展为患者。该患者 HSD17B3 基因发生致病突变引起 17β-HSD3 型缺陷症,导致雄烯二酮转化为睾酮障碍, 故表现为假两性畸形。
患者姐姐 (46XX) 虽然检测到两个杂合突变与患者相同,由于卵巢组织没有 17β-HSD3 型的表达,且女性的雄激素主要来自肾上腺,少量来源于卵巢,故睾酮合成和转化并不受影响,并不出现假两性畸形,临床可无任何表现或仅为月经紊乱,雄烯二酮和促性腺激素水平轻度增加等 [4-5],故患者姐姐能正常发育和生育。患者姐姐女儿染色体为正常 46XX,虽也携带有其中一个基因突变,但也并不会发展为患者。
17β-HSD3 型缺陷症治疗的主要目的在于患者性别的选择和男性假两性畸形的纠正。该患者及家属意见按女性性别生活,患者行双侧腹腔隐睾切除,肥大阴蒂切除,外阴整形术。术后予以补佳乐(戊酸雌二醇)1~2 mg/d 替代治疗。
参考文献
[1].Mendonca BB, Gomes NL, Costa EM, et al. 46,XY disorder of sex development (DSD) due to 17β-hydroxysteroid dehydrogenase type 3 deficiency[J].J Steroid Biochem Mol Biol, 2017,165(Pt A):79-85. DOI: 10.1016/j.jsbmb.2016.05.002.
[2]. Ben RB, Kallabi F, Mahfoudh N, et al. Novel cases of Tunisian patients with mutations in the gene encoding 17β-hydroxysteroid dehydrogenase type 3 and a founder effect [J].J Steroid Biochem Mol Biol,2017,165(Pt A):86-94. DOI: 10.1016/j.jsbmb.2016.03.007.
[3].Mendonca BB, Gomes NL, Costa EM, et al. 46,XY disorder of sex development (DSD) due to 17β-hydroxysteroid dehydrogenase type 3 deficiency[J].J Steroid Biochem Mol Biol, 2017,165(Pt A):79-85. DOI: 10.1016/j.jsbmb.2016.05.002.
[4].Rösler A, Silverstein S, Abeliovich D. A (R80Q) mutation in 17 beta-hydroxysteroid dehydrogenase type 3 gene among Arabs of Israel is associated with pseudohermaphroditism in males and normal asymptomatic females[J]. J Clin Endocrinol Metab, 1996, 81(5):1827-1831. DOI: 10.1210/jcem.81.5.8626842.
[5].Boehmer AL, Brinkmann AO, Sandkuijl LA, et al. 17Beta-hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations[J]. J Clin Endocrinol Metab, 1999,84(12):4713-4721. DOI: 10.1210/
更多精彩内容,关注「丁香智汇」